A research team led by Profs. JIANG Ling, ZHANG Zhaojun and ZHANG Donghui from the Dalian Institute of Chemical Physics (DICP) of the Chinese Academy of Sciences, in collaboration with Prof. KUO Jer-Lai from Institute of Atomic and Molecular Sciences, deconstructed vibrational motions on the potential energy surfaces of hydrogen-bonded complexes.
Their findings, published in CCS Chemistry, help to resolve the controversy of the exact vibrational assignment of each band feature of ammonia clusters.
Internal vibrations underlie transient structure formation, spectroscopy, and dynamics. However, at least two challenges exist when aiming to elucidate the contributions of vibrational motions on flexible potential energy surfaces (PESs). One is the acquisition of well-resolved experimental infrared spectra, and the other is the development of efficient theoretical methodologies that reliably predict band positions, relative intensities, and substructures.
Ammonia clusters are prototype systems for studying the interplay between different vibrational motions on flexible PESs. The researchers reported size-specific infrared spectra of ammonia clusters to address these two challenges.
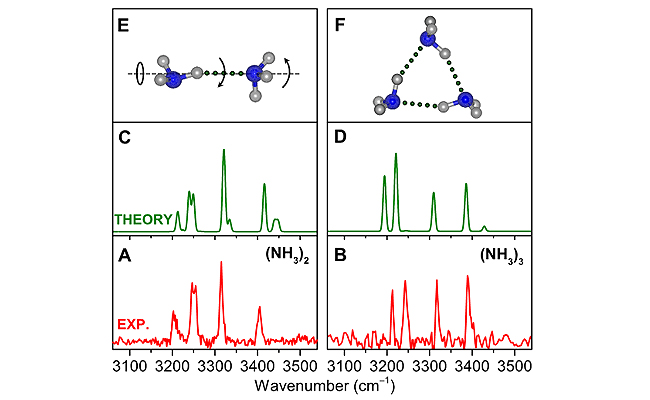
Comparison of experimental and theoretical IR spectra of (NH3)2 and (NH3)3 (Image by ZHANG Bingbing and YANG Shuo)
Unprecedented agreement between experiment and state-of-the-art quantum simulations revealed that the vibrational spectra were mainly contributed by proton-donor ammonia.
A striking Fermi resonance observed at approximately 3210 and 3250 cm-1 originates from the coupling of NH symmetric stretch fundamentals with overtones of free and hydrogen-bonded NH bending, respectively.
These findings contribute to a better understanding of vibrational motions in a large variety of hydrogen-bonded complexes with orders of magnitude improvements in spectral resolution, efficiency, and specificity. (Text by ZHANG Bingbing and YANG Shuo)