With the rapid development of biological macromolecules experimental technologies, more and more huge and complex molecular systems were found and identified, highlighting the unique multi-scale properties of biomolecules systems. Molecular dynamics simulation of biological molecules, as a powerful tool on identifying biomolecule functions, has become essential. However, due to the huge number of atoms contained in these systems, from thousands to millions, corresponding to the exercise of functions with time scales ranging from picoseconds to milliseconds, investigation of molecular and microscopic mechanism of their functions is very difficult.
Existing all-atom molecular simulation methods cannot be applied to such complex systems for high-precision and long-term dynamics simulations. Since returning in 2009, the molecular modeling and design research group has been focusing on the above issues. Based on the unique multi-scale features of biomolecular systems, we established mult-scale molecular modeling systems and carried out the development of simulation methods.
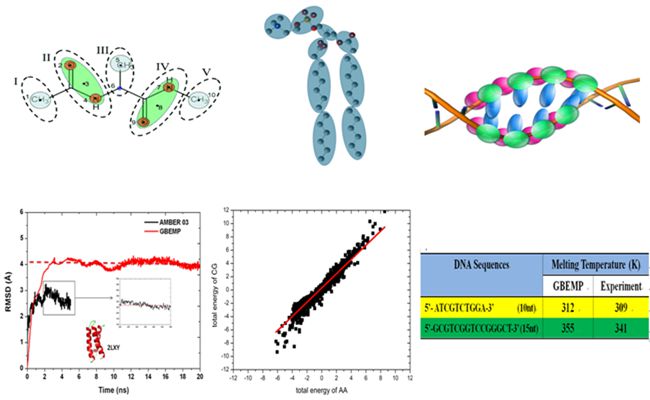
A Series of Progresses in Biomolecular Simulation Theory Development (Photo by LI Guohui)
By combining the high precision of all-atom force field functions as well as the fast calculation speed of coarse-grained models, we put forward an innovative development of Gay-Berne potential function, a generalized van der Waals potential that can be used to describe the interactions between non-spherical particles. The potential was combined with high-precision multi-polar distance expansion potential EMP and new high-precision coarse-grained molecular model GBEMP. The new force field was parameterized against 20 standard amino acids of proteins, DNA, RNA and cell membrane phospholipids and a series of biomolecular systems. With this high-accuracy coarse-grained model, molecular dynamics simulation of biological molecules achieved accurate results highly consistent with all-atom model, while the calculation speed is 10-100 times faster, thus achieved the optimal balance between accuracy and speed. J.Chem.Phys. 2011 (http://scitation.aip.org/content/aip/journal/jcp/135/15/10.1063/1.3651626),
J. Mol. Model. 2012 (http://link.springer.com/article/10.1007/s00894-012-1562-5),
J. Chem. Theory Comp. 2014 (http://pubs.acs.org/doi/abs/10.1021/ct400974z),
J. Comp. Chem. 2015 (http://onlinelibrary.wiley.com/doi/10.1002/jcc.23895/abstract),
J. Chem. Theory Comp.2016 http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00903.
Due to the high time-consumption of high-precision theoretical models, we proposed and developed an enhanced sampling algorithm correlated with GPU accelerated computing method, leading to an initiation of a high-precision, high efficiency simulation program, which is 5-10 times faster than previous work. The work was reported in the biomolecular simulation and free energy calculation conference in July, 2015, as the only invitee from mainland China. J. Comp. Chem 2016 (http://onlinelibrary.wiley.com/doi/10.1002/jcc.24227/abstract).(Text/Photo by LI Guohui)